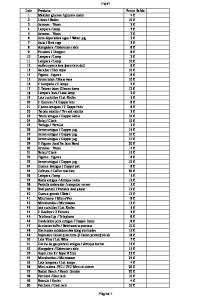
왗your lab focus 왘 (4) trained employees to follow the standard operating procedures manual. Linden and Kaplan4 emphasized that rapid identification of transfusion errors (especially AHTR) as well as properly identifying the cause of such errors and implementation of corrective actions are critical steps in the reduction of transfusion-associated fatalities. They cited previous work2 and recommended a number of strategies that may decrease the risk of AHTR. A modified version of these recommendations, including suggestions put forth by Sazama,1 is provided in [T2].
Conclusion Erroneous transfusion of ABO-incompatible blood is the most prevalent transfusion error and almost always reflects a preventable breakdown in transfusion protocol and standard operating procedure. These errors can have disastrous outcomes, accounting for significant iatrogenic morbidity and mortality. While most transfusion errors occur at the patient’s bedside and thus are remote from the blood bank and the immediate oversight of the transfusion service director, the hospital-wide implementation and enforcement of transfusion policies can
help minimize the risk and occurrence of transfusion errors.
1. Sazama K. Reports of 355 transfusion–associated deaths: 1976 through 1985. Transfusion. 1990;30:583-590. 2. Linden JV, Paul B, Dressler KP. A report of 104 transfusion errors in New York State. Transfusion. 1992;32:601-606. 3. Tourault MA, Mummert TB. Review of transfusion related fatalities: many preventable. Hosp Technol Scanner. 1993;11:1-3. 4. Linden JV, Kaplan HS. Transfusion errors: causes and effects. Transfus Med Rev. 1994;8:169-183.
CE update [cytology | hematology | generalist]
The Principles of Flow Cytometry Antony C. Bakke, PhD From the Department of Pathology, Oregon Health Sciences University, Portland, OR
왘
The uses of flow cytometry
왘
The basic components of flow cytometers
왘
Flow cytometry specimen analysis
왘
Flow cytometry quality control
Flow cytometry is defined as the simultaneous measurement of multiple physical characteristics of a single cell as the cell flows in suspension through a measuring device. It is a subset of the field of cytometry based on the use of specialized instruments. Flow Cytometry Compared With Other Techniques The original flow cytometer was the Coulter counter (Beckman Coulter M) invented by Wallace Coulter in the 1950s. All current automated, non–slide-based hematology instruments are flow cytometers. These instruments require cells in suspension and the ability to move cells
through the instrument. Blood is the ideal tissue because the cells are already in suspension and can be easily analyzed. The Coulter principle uses the resistance of a cell in an electrical current as it passes through an orifice to determine numbers of particles and the size of the particle. Flow cytometry has specifically come to denote the use of fluorescence measurement, usually with a laser light source. In laser flow cytometers, light scatter is used to measure the intrinsic size and granularity of the cell. In addition, fluorescence can be used to measure extrinsic features such as specific protein expression and nucleic acid content using added reagents, such as fluorescent stains and antibodies. Flow cytometers may be compared with fluorescence microscopes. With a microscope, the light source is a lamp, the objective lens focuses the image, and the eye is the detector. With a flow cytometer, the light source is a laser, an ob-
©
laboratorymedicine> april 2001> number 4> volume 32
jective lens focuses the light, and the detector is a photomultiplier tube [F1]. With a fluorescence microscope, the observer views cells and recognizes their different sizes. If a fluorescent probe is bound to the cell, a specific-colored fluorescence is emitted. Flow cytometry does not provide a focused image as does a microscope, but it does detect light and transform the signal into statistical data that can be presented in various graphic formats. Components of a Flow Cytometer Optics A laser beam is used in most flow cytometers as the light source to excite a fluorescent tag (eg, a fluorescent antibody) bound to the cells in the specimen. An argon laser, which produces blue light, is the most commonly used, although some instruments add a red helium-neon laser, and occasionally a mercury arc lamp is substituted. Before
Downloaded from http://labmed.oxfordjournals.org/ by guest on January 20, 2017
On completion of this article, the reader should be familiar with the basic components of a flow cytometer, the principles of its operation, specimen analysis, data presentation, and quality control. Cytology exam 0102 questions and the corresponding answer form are located after the “Your Lab Focus” section, p 213.
207
왗your lab focus 왘
1. Rapid analysis; rates of 50-4,000 cells/s 2. Total number analyzed can be very high; 5K100K+ (for rare events and accurate statistics) 3. Lack of information about intracellular probe distribution 4. Excellent quantitation of fluorescence per cell; range of 105× change 5. Single cell suspension critical 6. Correlated multiparameter analysis
1. Labor intensive; rates of 100 cells/min 2. Total number analyzed usually low, 1000 or fewer 3. Morphology and intracellular probe distribution; tissue sections 4. Quantilation of fluorescence limited; eye sensitivity approximately equal to 10× change
[F1] Light microscopy compared with flow cytometry for the detection of cells and cellular probes. Reproduced with permission from Becton-Dickinson M.
[F2] Light scatter measurements in a flow cytometer. The blue laser beam intersects the cell in the middle of the flow chamber. Forward scatter or forward angle light scatter (FALS) is measured along the axis of the laser beam. Blue laser light reflected off the cell and off internal structures in the cell is termed side scatter or 90-degree light scatter (90LS). Reproduced and altered with permission from Purdue University Cytometry Laboratories. The copyright for the original figure cannot be sold or transferred.
light, will fluoresce at a longer wavelength. Fluorescein gives a green fluorescence, phycoerythrin (an algal protein) gives orange, and peridinin chlorophyll protein or phycoerythrin coupled to cyanin 5 produces red fluorescence. Some flow cytometers have a second laser that can excite other fluorochromes. Like the side-scattered blue
laboratorymedicine> april 2001> number 4> volume 32
light, all of these fluorescent signals pass through the objective set at 90 degrees to the incident laser light. Additional dichroic mirrors reflect specific wavelengths of light, allowing separation of the signals from each fluorochrome with minimal overlap. In front of each photomultiplier, a final bandpass filter precisely defines the wavelength of light entering that detector.
©
Downloaded from http://labmed.oxfordjournals.org/ by guest on January 20, 2017
208
striking the cells, the light from either source passes through focusing and shaping optics to form an elliptical beam with a broad, maximal intensity in the center. The beam then passes through the center of a flow chamber, which is a quartz cuvette with an approximately 200-µm inner diameter. The specimen is also injected through the middle of this flow chamber, where the laser beam strikes the cell [F2]. When the laser strikes the cell, light is diffracted around the edges of the cell, producing a diffraction pattern along the path of the laser beam. This scattered light is approximately equivalent to the cell circumference and is the same wavelength as the exciting laser light (usually blue). Termed forward scatter (FSc) or forward angle light scatter (FALS), it is collected along the same axis as the laser beam. Because this is a very strong signal, a simple photodiode can convert the forward-scattered light into an electrical pulse. However, the direct laser beam could enter this photodiode, giving an overwhelming signal. A physical barrier eliminates the direct laser beam by blocking about a 2-degree angle of light along the path of the laser, allowing only the light diffracted at greater angles to pass. In addition to the forward-scattered light, blue laser light is also reflected off the cell and off internal structures in the cell. This is termed side scatter (SSc), right-angle light scatter (RALS), or 90degree light scatter (90LS) because it is detected by a collection objective set at 90 degrees to the incident laser light. It is a measure of granularity of the cell. Granulocytes have a much larger sidescattered light signal than do lymphocytes ([F1]; see also [F5]). After being focused by the objective lenses, the blue light is reflected by a dichroic mirror. These mirrors direct specific wavelengths of light into the correct detector. The detector is a photomultiplier tube, which converts the light signal into an electrical signal. Fluorescent probes can also be added to the cell for detecting specific molecules in the cell. These probes are typically antibodies to cellular antigens with a covalently attached fluorochrome. The fluorochrome, excited by the laser
왗your lab focus 왘 point of the laser, which is crucial for collecting accurate information about each cell in the specimen.
a
b
[F3] Flow cell showing hydrodynamic focusing of the specimen by the surrounding sheath fluid. Reproduced with permission from BectonDickinson M.
Specimen Analysis Specimen Preparation Specimens are often whole blood but may be other tissues. For analysis, the specimen must contain greater than 95% single cells for accurate interpretation of the data. Whole blood is a single cell suspension, but tissues must be disaggregated into single cells before analy-
c
Downloaded from http://labmed.oxfordjournals.org/ by guest on January 20, 2017
Fluidics The flow cytometer has 2 sources of fluid passing through it: a sheath fluid that is usually a buffered saline, and the specimen containing the cells to be analyzed. The specimen forms a core, which is injected into the middle of the sheath fluid, and both pass through the flow chamber [F3]. The outer sheath fluid causes hydrodynamic focusing, which compresses the specimen core into a small area. Cells are forced to pass singly through the focal
Electronics The photodiode detectors or photomultipler tubes first collect the light signal from the cells. In both cases, the photons produce a pulse of electrical current. Photomultiplier tubes have a voltage applied across a series of plates. This increases their sensitivity so that fewer photons are required to produce an electrical current. The initial small current requires amplification. The amplified current ranges from 0 to 10 V depending on the strength of the signal. The voltage signal is an analog signal and cannot be recognized by the computer. It is converted by an analog-todigital converter into a digital signal, called a channel, which can range from 0 to 1,023 channels. The channel is a counter, and all signals of a specific voltage are counted in a channel corresponding to the strength of the signal. The amplification of the initial signal can be either linear or logarithmic. Linear amplification provides a direct visual relationship and is useful for scatter signals and fluorescent measurements of DNA. Linear amplification allows a dynamic range of approximately 200- to 1,000-fold. Logarithmic amplification is used for most other biologic signals, including immunofluorescence, owing to
the extreme dynamic range of these signals. Logarithmic amplification gives a dynamic range of 2,000- to 10,000-fold. The fluorescent signals have been discussed above. Although these signals appear mainly as a single color under the microscope, actually the emission from any fluorochrome is broad. Although fluorescein isothiocyanate (FITC) appears apple-green, it also has lower-level emissions in the orange area of the spectrum. Hence, any green FITC-stained cell will also generate a signal in the orange channel, making the cell appear to be labeled by 2 fluorochromes. To allow multicolor fluorescence, this apparent overlap must be eliminated. This is done by fluorescence compensation. A percentage of any bright signal, such as FITC, is subtracted from any other associated signal. The instrument is set up at the beginning of a run with pure signals of each fluorochrome, and any associated fluorescence at the other wavelengths is compensated [F4].
209
[F4] Electronic compensation corrects spectral emission overlaps. Two-parameter contour plots of fluorescein vs phycoerythrin fluorescence. a, No compensation produces results with many cells apparently expressing both fluorochromes, although these are mutually exclusive stains (CD4 and CD8). b, Overcompensation leaves many cells on the axes and may decrease dual expression when present. c, Correct compensation produces results in which mutually exclusive stains appear to be pure green or orange and few dual-labeling cells are present. FL1-H, x-axis, green; FL2-H, y-axis, orange.
©
laboratorymedicine> april 2001> number 4> volume 32
왗your lab focus 왘 a
b
c
0%
24%
Ungated total particles M1
Gated lymphocytes
11% 65%
100
101
102
103
FL1-H
[F5] Data can be displayed on several plots and electronically gated to analyze pure populations. a, Dot plot of lysed whole blood showing forward scatter vs side scatter with a gate surrounding lymphocytes. b, Overlaid histograms of fluorescein fluorescence showing the difference with total and gated population analysis. The positive cells within the markers are 1% of the total particles, including debris, but 89% of the gated lymphocytes. c, Dot plot of fluorescein vs phycoerythrin fluorescence showing quadrant statistics. FL1-H, x-axis, green, fluorescein; FL2-H, y-axis, orange, phycoerythrin.
210
Data Processing The initial light signal is first converted by the detectors into an electric current, which is amplified and measured as an analog quantity, such as 5 V of a maximum of 10 V. Because this signal is analog, it must be converted into a digital signal for computer processing. The signal then becomes an event with a specific digital channel value such 512 on a 1,024-channel scale. The digital data can be displayed as 1-parameter histograms [F5b] or as 2-parameter plots [F4, F5a, F5c]. The information on a 1-parameter histogram is described either as the per-
centage of cells within a set of markers or as the mean fluorescence intensity of a population. For logarithmically amplified signals, the geometric mean fluorescence is used, since it is less influenced by the extreme ends of the distribution. A 2parameter plot is usually divided into 4 quadrants, each containing a percentage of the total population [F5c]. This division is used to distinguish between fluorescent and nonfluorescent cells. It also defines expression and nonexpression of a cell molecule marked by a fluorescent antibody or other fluorochrome. Because the specimen that is analyzed may be complex with several populations plus debris, such as lysed whole blood, analysis of the total specimen would give variable and confusing results. Therefore, it is often necessary to analyze a single population, such as lymphocytes. Electronic gating allows separation of the total population into parts, which can be individually analyzed [F5a]. Quality Control There are several kinds of quality controls. First the flow cytometer itself must be evaluated for proper function. This is usually accomplished with standardized fluorescent beads. These give very precise, reproducible patterns, which quickly assess instrument func-
laboratorymedicine> april 2001> number 4> volume 32
©
tion. In addition, the beads are fairly stable and can be used daily with little change in values. The bead values for instrument settings and signal intensity are used to generate Levy-Jennings plots and determine instrument errors. A second quality control material is used to set up the appropriate instrument settings for the type of staining used. These can be beads or antibody-stained cells. One of the primary functions of this second control is to establish the compensation settings for the instrument; therefore, either beads or cells should mimic the staining that will be used on actual specimens. The third level of quality control is a control substance that mimics actual specimens. These controls are available commercially and usually consist of stabilized blood, sometimes with added tissue-culture cells that mimic a specific cancer cell. These controls can be used to assess the sensitivity of the instrument. They will have published ranges for many of the antibodies used in the laboratory. Because they usually contain normal, stabilized blood cells, some proteins characteristic of specific cancer cells will not be present. For this reason, some tissue-culture cells may be included because they express specific proteins that can be used to evaluate other antibodies.
Downloaded from http://labmed.oxfordjournals.org/ by guest on January 20, 2017
sis. Often antibodies are used to stain cells and select subpopulations or evaluate protein expression. In most cases, these antibodies are commercial products with robust staining properties. However, the staining patterns should be confirmed in the laboratory on each new lot of antibody using appropriate control materials. Unknown antibodies must be carefully evaluated for specificity and efficacy of staining. This is best done under a microscope initially because nonspecific staining or unusual patterns can be better distinguished with a microscope than with a flow cytometer. Once approximate staining characteristics are determined, the procedure can be fine-tuned for the flow cytometer.
Conclusion Flow cytometry is used in the clinical laboratory for a variety of analyses including diagnostic immunophenotyping of lymphomas, leukemias, and congenital immunodeficiencies; detection of minimal residual disease in leukemias; monitoring CD4+ T-lymphocyte counts in HIV patients; DNA content analysis for prognosis of malignancies; screening of hematologic disorders such as paroxysmal nocturnal hemoglobinuria and idiopathic thrombocytopenic purpura; evaluation of peripheral stem cell products for transplantation; monitoring monoclonal antibody
therapy; and determination of the extent of maternal/fetal bleeding by detection of hemoglobin F. Flow cytometry remains a laborintensive area of laboratory medicine requiring extensive training. Learning the fundamental principles of flow cytometry enhances both patient care and work satisfaction. In addition, these principles apply to other areas using cytometry techniques, such as hematology. For more details and theoretical discussion, the reader is referred to several books 1-4 and Internet sites.5,6
1. Givan AL. Flow Cytometry: First Principles. New York, NY: John Wiley & Sons; 1992. 2. Bauer KD, Duque RE, Shankey TV, eds. Clinical Flow Cytometry: Principles and Application. Baltimore, MD: Williams and Wilkins, 1993. 3. Owens MA, Loken MR. Flow Cytometry Principles for Clinical Laboratory Practice, Quality Assurance for Quantitative Immunophenotyping. New York, NY: Wiley-Liss; 1995. 4. Melamed MR, Lindmo T, Mendelsohn ML. Flow Cytometry and Sorting. 2nd ed. New York, NY: Wiley-Liss; 1990. 5. Purdue University Cytometry Laboratories. http://flowcyt.cyto.purdue.edu/. Accessed January 8, 2001. 6. Hematopathologic phenotypes made mockingly simple. Available at: http://www.neosoft.com/ ~uthman/cdphobia/cdphobia.html. Accessed January 8, 2001.
Downloaded from http://labmed.oxfordjournals.org/ by guest on January 20, 2017
PostScript Picture
211
laboratorymedicine> april 2001> number 4> volume 32